IEC 60601-1-2 Testing Guide for Medical Device EMC
What is IEC 60601-1-2 and what is required to EMC test my medical device so I can submit my product for approval medical regulators?
As one of the leading IEC 60601-1-2 testing labs, Sunfire Testing has extensive experience working in the voluminous IEC 60601 standards, and specializes in EMC testing to the IEC 60601-1-2, 4th Edition.
Chances are you have not typed IEC 60601-1-2 by mistake and you have an actual medical product (or an idea for medical product), and are trying to determine what type of EMC testing is required in order for your product to be approved by end regulators.
In this guide, we will at the cursory level examine IEC 60601, discuss how IEC 60601 dovetails into the EMC testing required for IEC 60601-1-2, and then go straight into an analysis of IEC 60601-1-2, 4th edition and explain what is required for the testing.
Who created the IEC 60601 standard in the first place?
![]()
As with all CE Testing; the original authors of the IEC 60601 standards is the International Electrotechnical Commission, which is commonly referred to as IEC.
The IEC was founded in Europe in 1906, and currently has 89 member countries across the world. While the IEC is based in Geneva Switzerland, membership extends across the world and the IEC standards form the basis of all product safety testing.
For purposes of medical device testing, IEC 60601, is one of hundreds of family standards the IEC has developed for safety testing different types of products.
From a global perspective, every major medical device market of the world expects testing of your end product to IEC 60601. The only major variation across the world is which specific versions of IEC 60601 standard is accepted in a specific country.
How long has IEC 60601 been around?
The first edition of IEC 60601 was created in 1977, and has been a true work in progress from the IEC since then.
The entire IEC 60601 standards, in its current iteration, stands with the general standard, 10 major collateral standards, and around 80 particular standards.
If you were to print out all of the standards which make up the IEC 60601 family, you are likely in the over 10,000 pages category.
As we said earlier, IEC 60601 is by far the largest standard and family of related standards that the IEC publishes. It is not an easy set of documents to work with.
(Fun fact, we also would guess purchasing the entire series of the standard probably is also close to $10,000 USD – $1/page. You could not hire us to write one page of how dense the standard is for $1/page either!).
In fairness though to the IEC ensuring medical products are safe for use is quite a task as well.
Why is IEC 60601 so difficult to work with and understand?
In terms of complexity of testing and regulatory verbosity, the IEC 60601 standards towers above any another IEC standard that is currently published. As a product manufacturer, you are not alone in in trying to tease out the nuts and bolts of what is required for testing your medical product.
Much of the difficulty in dealing with the IEC 60601 standards relates back to the subject matter and the approach taken by the IEC subcommittee who drafted the standards.
From a breadth of product perspective, there is no singular “medical device” which functions like a toaster oven and the enormity of functional variation is really almost infinite.
In terms of having to use language to describe and encapsulate all the possible devices which could qualify and are used for “medical” purposes, the actual standards need to be written in a certain tone and purpose which leave them generic enough to be applied to a wide range of products.
The generality of the IEC 60601 standards, is really the best way to understand the idea of “Risk Management” [which is a huge buzzword, as you work through the IEC 60601 standards].
The entire goal of IEC 60601 is to manage medical device risk. Put loosely, the goal is not to remove all possible risks and demonstrate through testing that no risk could exist – the goal is to remove as much risk as possible and still be economically profitable and viable as a product.
We also agree, the concept of removing “as much risk” as possible is hopelessly undefined from an engineering perspective. For the teams of designers & quality standards who are left with reviewing the guidelines of ISO 14971 and coming up with a Risk Management File, we can certainly sympathize.
As we work through IEC 60601 and lead into IEC 60601-1-2 testing, just keep in your mind the regulatory goal of the standard: minimizing risk, but not completely eliminating.
Once you are able to get into this regulatory mindset, what you will find is that IEC 60601-1-2, 4th Edition is actually pretty reasonable and approachable in the greater context of IEC 60601-1-2.
You mentioned collateral and particular standards earlier, how do those connect to moving from IEC 60601 to the EMC testing I am required to perform under IEC 60601-1-2?
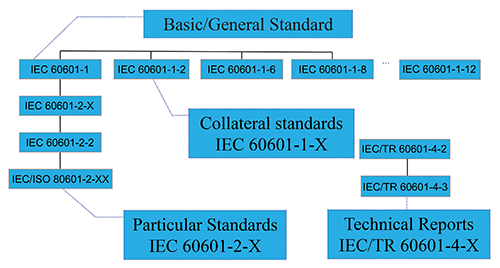
In the image above, you will see a descriptive break out of how the IEC committee has decided to delineate the IEC 60601 family of standards.
This graphic is a little confusing, but the organization of the standard actually is pretty logical.
At the top of the hierarchy is the IEC 60601 standard itself. This is the central operating document of the entire process. If you are working on a medical device, you will have spent the majority of your time reviewing IEC 60601 and fulfilling the requirements of the standard.
There are then two different categories of “sub” standards. These are the so-called collateral standards and particular standards.
The way to think about a collateral standard (which is noted as a -1-X, [the X is final number of the collateral standard]), is a high level regulatory requirement which applies to almost all medical devices which are tested to IEC 60601.
In a sense the collateral standards, would be better referred to as, “related” standards or something a little more descriptive, but just remember that any -1 standard you see is a standard which applies to almost all medical products.
The list of IEC 60601 Collateral Standards is much shorter than the particular standards and include:
- IEC 60601-1-2, Part 1-2: General requirements for basic safety and essential performance - Collateral Standard: Electromagnetic disturbances - Requirements and tests
- IEC 60601-1-3, Part 1-3: General requirements for basic safety and essential performance - Collateral Standard: Radiation protection in diagnostic X-ray equipment
- IEC 60601-1-6, Part 1-6: General requirements for basic safety and essential performance Collateral standard: Usability
- IEC 60601-1-8, Part 1-8: General requirements for basic safety and essential performance - Collateral Standard: General requirements, tests and guidance for alarm systems in medical electrical equipment and medical electrical systems
- IEC 60601-1-9, General requirements for basic safety and essential performance - Collateral Standard: Requirements for environmentally conscious design
- IEC 60601-1-10, General requirements for basic safety and essential performance - Collateral Standard: Requirements for the development of physiologic closed-loop controllers
- IEC 60601-1-11, General requirements for basic safety and essential performance - Collateral Standard: Requirements for medical electrical equipment and medical electrical systems used in the home healthcare environment
- IEC 60601-1-12, General requirements for basic safety and essential performance - Collateral Standard: Requirements for medical electrical equipment and medical electrical systems intended for use in the emergency medical services environment
So we have a list of 8 collateral standards, which are general high level standards which are applied to common medical devices. Did you notice that IEC 60601-1-2 is one of these so called collateral standards?
From the context of IEC 60601, EMC testing a medical device is considered almost perfunctory for any conceivable medical device and the IEC decided that such EMC testing would then be codified in a collateral standard and they randomly decided to call that EMC testing standard IEC 60601-1-2.
It’s really that simple.
Similarly, if you examine the titles or over the course of your IEC 60601 project you’ll find that the other collateral standards are similarly fairly common and high level testing requirements that you expect to see in a typical medical device.
You see, the folks at the IEC do have a bit of a game plan in place here for understanding how to navigate the standards.
The second category of standards is the so called particular standards. There are nearly 80 particular standards, but for just a sampling of some of them and to understand what they are:
- IEC 60601-2-20, Particular requirements for the basic safety and essential performance of infant transport incubators
- IEC 60601-2-21, Particular requirements for the basic safety and essential performance of infant radiant warmers
- IEC 60601-2-22, Particular requirements for basic safety and essential performance of surgical, cosmetic, therapeutic and diagnostic laser equipment
- IEC 60601-2-23, Particular requirements for the basic safety and essential performance of transcutaneous partial pressure monitoring equipment
- IEC 60601-2-24, Particular requirements for the basic safety and essential performance of infusion pumps and controllers
- IEC 60601-2-25, Particular requirements for the basic safety and essential performance of electrocardiographs
- IEC 60601-2-26, Particular requirements for the basic safety and essential performance of electroencephalographs
- IEC 60601-2-27, Particular requirements for the basic safety and essential performance of electrocardiographic monitoring equipment
- IEC 60601-2-28, Particular requirements for the basic safety and essential performance of X-ray tube assemblies for medical diagnosis
- IEC 60601-2-29, Particular requirements for the basic safety and essential performance of radiotherapy simulators
- IEC 60601-2-31, Particular requirements for the basic safety and essential performance of external cardiac pacemakers with internal power source
(Check here for a full list of the IEC 60601 particular standards.)
Do you notice that the standard descriptions are much more specific to a type of product? This is the “particular” element that the IEC was trying to impart.
The way to think about a particular standard (which is noted as a -2-X, [the X is final number of the particular standard]), is a specific regulatory requirement which applies to the “type” of medical device that you are manufacturing.
A particular standard usually overrides the standard requirements in the main IEC 60601 standard or any collateral standards. What that means is if you examine a particular standard you will find that it may say something to the effect of “instead of applying clause A.B.C of IEC 60601-A-B, use these limits or rules as found in this standard instead”.
As you can imagine, while 80 particular standards which specify a specific type of equipment sounds like a lot on a paper, sometimes you may have a novel medical device which does not so clearly fit into one particular standard and there is much discussion and justification that follows in then determining and justifying which standards your product needs to be evaluated against.
For the purposes of EMC testing a medical device, IEC 60601-1-2, however does not have any real ambiguity. If you are producing a electronic medical device, at a minimum you will require testing to IEC 60601 and IEC 60601-1-2.
What is the current version of IEC 60601-1-2, this guide is based on?
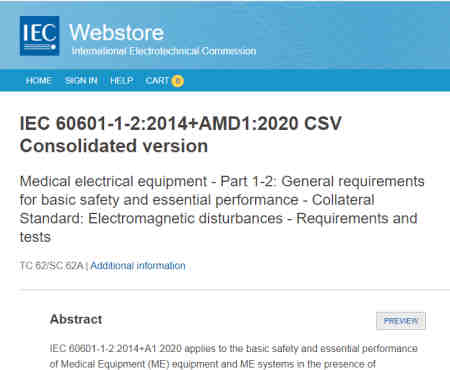
As of 2021, the most current version of IEC is IEC 60601-1-2:2014+AMD1:2020 CSV. Over time, you should expect to see future amendments and revisions to IEC 60601-1-2.
Generally speaking, the updates over time are usually clarification and new guidance on the core standards and don’t chance the required testing in a major way.
IEC 60601-1-2:2014 is commonly called the 4th Edition. For purposes of our analysis we will be discussing on the 4th edition only; however, depending on which country you are seeking approval in you may be required to test earlier versions.
So IEC 60601-1-2 is a collateral standard of IEC 60601, and my medical device requires EMC testing. What does this involve?
Now that we have a little bit of context to IEC 60601 family of standards, and understand the connection to IEC 60601-1-2 (and hopefully a little bit better understanding of how the medical device EMC testing connects back to the other IEC 60601 testing), we can now dive into the actual IEC 60601-1-2 standard and see what is required.
What exactly is IEC 60601-1-2 and how does it differ from other CE EMC testing standards?
If you have ever been through CE testing, to a standard like EN 55032 or EN 55035 for IT & AV equipment], or tested laboratory / measurement equipment to EN 61326-1 you will find that the EMC testing for IEC 60601-1-2 is quite similar in the overall approach.
The EMC testing in both EN55032/EN55035 and EN 61326-1 both have two major elements: emissions testing and immunity testing.
The Emissions Testing as per IEC 60601-1-2 involve four possible emissions measurements:
- Conducted RF Emissions (CISPR 11:2009+A1:2010)
- Radiated RF Emissions (CISPR 11:2009+A1:2010)
- Harmonic Distortion (IEC 61000-3-2:2005+A1:2008+A2:2009)
- Voltage Fluctuation and Flicker (IEC 61000-3-3:2013)
The Immunity Testing as per IEC 60601-1-2 involve 7 possible immunity tests:
- Electrostatic Discharges (IEC 61000-4-2:2008)
- Radiated RF EM Fields and Proximity Wireless fields (IEC 61000-4-3:2016+A1:2007+A2:2010)
- Electrical Fast Transients and bursts (IEC 61000-4-4:2012)
- Surges (IEC 61000-4-5:2005)
- Conducted Disturbances, induced by RF fields (IEC 61000-4-6:2013)
- Voltage Dips and Interruptions (IEC 61000-4-11:2004)
- Rated Power-frequency Magnetic Field (IEC 61000-4-8:2009)
What are the four emissions measurements of IEC 60601-1-2?
1) Conducted RF Emissions (CISPR 11:2009+A1:2010) 
CISPR 11 is a very common EMC standard which is used to measured the conducted emissions of a product. The conducted emissions of a product are the EMI output of the device which is then measured at the AC mains. Essentially this is a measurement of the noise coming out of your medical device and flowing back into the AC mains.
2) Radiated RF Emissions (CISPR 11:2009+A1:2010) 
Radiated RF emissions is the classic EMC measurement and the most fundamental EMI concept. IEC 60601-1-2 measures the emissions of your device which are unintentionally radiated from the electronics of your device out into the open air.
3) Harmonic Distortion (IEC 61000-3-2:2005+A1:2008+A2:2009) 
IEC 61000-3-2 is a measurement that aims to reduce the harmonic currents which flow back into the AC mains which a by-product of a power supply taking sinusoidal AC mains voltage and then “smoothing” out the AC into DC power (typically with capacitors). If the power supply of your product is not designed correctly it will produce too much harmonic back-feed into the AC mains and this can affect other devices which are sharing the same mains power lines.
4) Voltage Fluctuation and Flicker (IEC 61000-3-3:2013) 
Voltage Fluctuation and Flicker, as per IEC 61000-3-3, is a test to prove that when your medical device is connected to the shared AC mains the current drawn from the device (and related power supply) will not cause other connected devices on the AC mains to “flicker” or “fluctuate”. [If you remember seeing your old tube TV flicker when the vacuum is connected to the AC mains, this is the phenomenon IEC 61000-3-3 is trying to prevent).
What are the eight immunity tests of IEC 60601-1-2?
1) Electrostatic Discharges (IEC 61000-4-2:2008) 
ESD Testing, under IEC 60601-1-2 is likely the most difficult measurement of the process. Electrostatic Discharge, as per IEC 61000-4-2:2008, is a test of “zapping” your medical device with simulated electrostatic pulses (like when you walk over carpet and touch a metallic object). Unlike with consumer product tested to other CE limits, medical devices are required to be significantly more immune to electrostatic discharge.
2) Radiated RF EM Fields and Proximity Wireless fields (IEC 61000-4-3:2016+A1:2007+A2:2010)
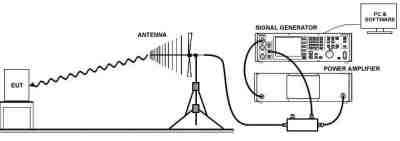
IEC 61000-4-3, is a test of your medical device to demonstrate that it will continue to function as intended in the presence of other device’s EMI emissions and RF emissions. In essence, this is a test to demonstrate that when your medical device is placed next to another device which has emissions, your device will function correctly.
3) Electrical Fast Transients and bursts (IEC 61000-4-4:2012) 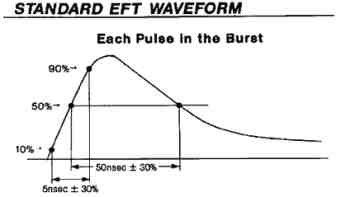
Electrical Fast Transients and bursts, commonly referred to as EFT testing, is a simulated test where the AC mains are setup to have simulated bursts of noise which are then fed into your medical device. IEC 61000-4-4 requires that under the EFT testing your device does not malfunction or latch up when the EFT bursts are fed into the device.
4) Surges (IEC 61000-4-5:2005)
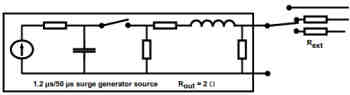
Similar to the EFT testing of IEC 61000-4-4, Surge testing under IEC 61000-4-5 is another simulated AC mains event however in this case the simulated test case is when there is a surge voltage event which could be a lightning strike which then injects high currents into the power mains. Your medical device will be tested to these simulated high voltage events and ensure that it will not fail during an event.
5) Conducted Disturbances, induced by RF fields (IEC 61000-4-6:2013) 
IEC 61000-4-6, is a similar measurement of RF immunity as is performed in IEC 61000-4-3. In IEC 61000-4-3, your device is exposed to outside emissions which are transmitted via external antennas to simulated RF / EMI noise that other nearby devices could be producing. These same sorts of RF / EMI noise are re-used but under IEC 61000-4-6, the cabling of your device is exposed to these sources of noise instead of your core device. Your cabling is expected to be immune to these sorts of noise as well as the core device.
6) Voltage Dips and Interruptions (IEC 61000-4-11:2004)

Voltage Dips & Interrupts, under IEC 61000-4-11 are quite similar EFT & Surge Testing. Under this requirement, your device is exposed to short duration dips and variations in voltage in the supplied AC power. Your medical device is expected to be able to function correctly and accurately when these short term disruptions of voltage are applied.
7) Rated Power-frequency Magnetic Field (IEC 61000-4-8:2009) 
As the final required immunity test, IEC 61000-4-8, is related to IEC 61000-4-3. Under IEC 61000-4-3, an external antenna is used to transmit RF / EMI noise at your medical device and ensure that it functions correctly. IEC 61000-4-8 replaces RF / EMI noise with simulated magnetic fields. Your product is evaluated similarly but instead of simulated RF / EMI, it is expected to be able to operate in the presence of common magnetic field ranges which are by products of AC mains frequencies.
It seems like a lot of the medical device testing relates to the immunity testing; how do we define and prove our medical device is not susceptible to this immunity testing?
As IEC 60601 has evolved as a standard the concept of the how to prove your medical device will operate as intended when it experiences outside electro-phenomena (as those are simulated during immunity testing) has continued to be of great difficulty and much discussion.
When performing safety testing to IEC 60601, a device manufacturer is expected to apply the concept of Risk Management (which is based upon ISO 14971) and create a formal system called the Risk Management File (RMF) which is used to catalog the risks of the medical, details controls and decision changes used to control such risks, and then catalog the likelihood of such failures occurring and demonstrating that risk has been minimized as much is economically viable.
At the core of the Risk Management process, a medical device manufacturer is called upon to determine the Essential Performance of a device.
Essential Performance is the bedrock starting point of catalog all perceived risk of the device. The thought exercise that a medical device manufacturer is called upon is to lay out what the medical device is expected to do 100% of the time. In essence, Essential Performance is the absolute performance requirements of the medical system.
Under ISO 14971, once Essential Performance is defined a device manufacturer is called upon to define the Basic Safety of the system (which at a high level is the associated controls and design decisions implemented in order to ensure the Essential Performance of the device is provided at all times).
Understandably, the Risk Management process under ISO 14971 creates a very broad framework to work from. Under the process, a device manufacturer is expected to interpret and provide a full justification of how and why their system is safe.
Because the exercise is so open ended, this leads to a lot of difficulties in creating a Risk Management File; and in then performing the underlying IEC 60601 safety testing.
However, under IEC 60601-1-2 the determination of how to define if your medical device is passing the EMC testing is much more straightforward and technical as opposed to the more open ended Risk Management process under ISO 14971.
Pass/ Fail Criteria under IEC 60601-1-2
The assumption under IEC 60601-1-2 is that as a medical device manufacturer you have considered and calculated the potential failure cases of your device and have “included the possible effects of Electromagnetic Disturbance”.
What this means is that in your Risk Management Files, as the designer of your device you have considered the IEC 61000 testing which is going to be performed in IEC 60601-1-2 and have some ideas as to how and what a failure of your device would look like in the context of it’s Essential Performance (and hopefully you’ve designed some protective measures into your device to maintain Basic Safety of the device – see how this works?).
Under IEC 60601-1-2, there is a defined concept refereed to as the Immunity pass/fail criteria.
Pass/fail criteria are intended to be a device manufacturer’s specification of what the failure of their medical device would be based on the following elements (IEC 60601-1-2, Annex I, I.2.3):
- the HAZARDS
- the functions to be tested for IMMUNITY to verify freedom from unacceptable RISK
- the criteria on which to base the pass/fail decision
- operating modes
- characteristics of simulated PATIENT physiological signals
- specification of locations of INTENDED USE
- the characteristics of the test, where these are at the discretion of the MANUFACTURER
Once again we are left with some high level language for IEC 60601, but the concept is pretty straightforward. Based on the specifics of your medical device, what are reasonable ways in which you would say that the device has failed.
IEC 60601-1-2, Annex I, I.3, some common examples of pass/fail criteria are provided:
- malfunction
- non-operation when operation is required
- unwanted operation when no operation is required
- deviation from normal operation that poses an unacceptable RISK to the PATIENT or OPERATOR
- component failures
- change in programmable parameters
- reset to factory defaults (MANUFACTURER’s presets)
- change of operating mode
- a FALSE POSITIVE ALARM CONDITION
- a FALSE NEGATIVE ALARM CONDITION (failure to alarm)
- cessation or interruption of any intended operation, even if accompanied by an ALARM SIGNAL
- initiation of any unintended operation, including unintended or uncontrolled motion, even if accompanied by an ALARM SIGNAL
- error of a displayed numerical value sufficiently large to affect diagnosis or treatment
- noise on a waveform in which the noise would interfere with diagnosis, treatment or monitoring
- artefact or distortion in an image in which the artefact would interfere with diagnosis, treatment or monitoring
- failure of automatic diagnosis or treatment ME
As you can see, the potential pass/fail criteria are pretty generalized but in the context of Essential Performance are fairly reasonable indicators of a device failure.
The next step is to provide your test lab with a formal document called a Test Plan which outlines the manufacturer’s determination of the pass/fail criteria and spell out how to evaluate the performance of the device while it is under immunity testing.
Before we move onto the Test Plan, what about the 4 emissions tests?
In terms of the emissions testing IEC 60601-1-2, those measurements are fairly discrete and self contained.
What we mean by that, is that the only variable and discussion point on measuring the emissions of a medical device is to ensure that is it is being “fully exercised” and that operating modes of the device have been fully evaluated.
Because we are not really considering any element of the Risk Management process, these measurements do not require as much formal documentation and analysis, and it’s really a turn-on-the-device and measure sort of process.
That being said, the Test Plan and discussions leading up to the testing will include operational details of the device and how to best operate it to its fullest extent.
What is required in an IEC 60601-1-2 Test Plan?
Under IEC 60601-1-2, there is a formal stated requirement that a test lab be provided with a formal document which outlines the following elements of your medical device (IEC 60601-1-2; Annex G):
- Name and address of the test facility
- Description of the ME EQUIPMENT or ME SYSTEM
- Description of the BASIC SAFETY and ESSENTIAL PERFORMANCE including a description how the BASIC SAFETY and ESSENTIAL PERFORMANCE will be monitored against the pass/fail criteria during each test
- Identification of the ME EQUIPMENT or ME SYSTEM
- ME EQUIPMENT or ME SYSTEM software / firmware version of the sample to be tested
- Number of samples to be tested The number of samples for each EMC test
- INTENDED USE and intended environments
- Applicable standards and test methods A list of the standards (with dates) and EMISSIONS limits or IMMUNITY TEST LEVELS
- Deviations from the Basic EMC standards
- Applicability / tests that will not be performed The decision and justification not to perform a measurement or test shall be documented.
- If the procedure specified by Annex E or an equivalent procedure is used ; a justification for any SPECIAL ENVIRONMENTS identified or adjustments made; the adjusted reasonably foreseeable maximum EM DISTURBANCE levels; – the resulting final IMMUNITY TEST LEVELS, rounded to the nearest whole number or, if a decimal, to a single significant digit; details of the methods and data sources used in determining the appropriate IMMUNITY TEST LEVELS
- IMMUNITY TEST LEVELS for each IMMUNITY test and EMISSIONS compliance class and group
- IMMUNITY pass/fail criteria Specific IMMUNITY pass/fail criteria for BASIC SAFETY and ESSENTIAL PERFORMANCE as per the RISK ANALYSIS (see Annex I)
- ME EQUIPMENT or ME SYSTEM configurations, settings and operating modes
- Test setup electrical and physical diagrams S
- ME EQUIPMENT or ME SYSTEM power input voltages and frequencies
- Earthing configuration
- Whether the ME EQUIPMENT or ME SYSTEM will be tested as table-top or floor-standing equipment, or a combination of the two
- Testing of PERMANENTLY INSTALLED LARGE ME EQUIPMENT or LARGE ME SYSTEM
- Exercising of SIP/SOPS
- For floor-standing ME EQUIPMENT or ME SYSTEMS, the height of the support
- Description of any PATIENT-COUPLED cable terminations to be used
- Simulators, accessories and auxiliary equipment Describe simulators, ACCESSORIES and auxiliary equipment used, including PATIENT physiological and subsystem simulation
- Documentation of any special ME EQUIPMENT or ME SYSTEM hardware or software needed to perform the tests
- ALARM LIMIT settings If applicable, provide rationale for the settings chosen.
- Planned ESD test points. If possible, include a drawing or annotated photo showing the ESD test points.
- Dwell time for each IMMUNITY test requiring a dwell time
As you can see this quite an extensive list and in practice at Sunfire Testing we will help work with your team and identify the elements of the test plan and documentation that we will need in order to perform your IEC 60601-1-2 testing.
Once I have a test plan and have worked with Sunfire Testing on the planning; what else is left to run and complete the IEC 60601-1-2 testing?
Congratulations! By the time we have devices in hand and have worked with you to plan on what is required for the IEC 60601-1-2 testing, you have gone through and and spent quite some time considering the Essential Performance of your device and formulated what are reasonable pass fail criteria.
This is no small feat and is what separates IEC 60601-1-2 testing from more typical CE consumer electronics testing. With this documentation in hand, we can then begin the formal chamber testing of your medical device and then provide testing reports.
As part of IEC 60601-1-2, there are a number of requirements in your product’s Instrutions for Use and Labeling. While we are working on the formal chamber testing of your medical device we will assist in the documentation compliance elements of your medical device and ensure that it meets all the “soft” compliance elements of IEC 60601-1-2 as well.
At the end of the testing process, you will receive a test report which documents the requirements of IEC 60601-1-2 and outlines what testing is required.
How can Sunfire Testing help us with our IEC 60601-1-2 Testing?
Part of what makes IEC 60601-1-2 testing difficult is not primarily the EMC measurements, but it is adapting to the language and thought process of IEC 60601 and applying the concepts of Risk Management under ISO 14971 and then applying those concepts as it relates to the EMC testing of your medical device.
As former product designers, we are unique in our ability to discuss your medical device in the context of EMC testing but also on a design level. By being able to discuss your medical device at a design level, we are able to assist in the Test Plan discussions and immunity pass/fail criteria at a much higher level then a typical testing lab will be able too.
We’ve found that this greatly speeds up the quoting process and we are able to get our client IEC 60601-1-2 testing projects underway at vastly accelerated speeds.
Our IEC 60601-1-2 testing clients range from established medical companies to startup entities producing a novel medical device, and no single IEC 60601-1-2 testing product or project is the same. Your lab’s ability and background knowledge in medical devices plays a huge role in the success and speed of your testing.
If you are looking for IEC 60601-1-2 testing or have any questions on your next medical project, contact us for a free quote and analysis of the required IEC 60601-1-2 testing.
Our typical IEC 60601-1-2 testing project fees start from $6999 and include free re-testing and assistance on the documentation and labeling of your medical device to fully comply with IEC 60601-1-2.



We make compliance testing easy.
Submit your project details today for a no-cost quote
Request Quote